Much has been written about the potential of Artificial Intelligence (A.I.) ever since John McCarthy proposed the term in 1955. The idea, initially carried forward through science fiction, gained commercial interest towards the end of the last century. Today, there are literally hundreds of applications for A.I. in medical devices alone.
But there is a fundamental problem with A.I. nicely summed up in Gartner’s thought-provoking Hype Cycle for Artificial Intelligence, 2021[i]. While there is clearly a lot of promise for A.I., it seems that, thus far, A.I. often delivers only that … promises.
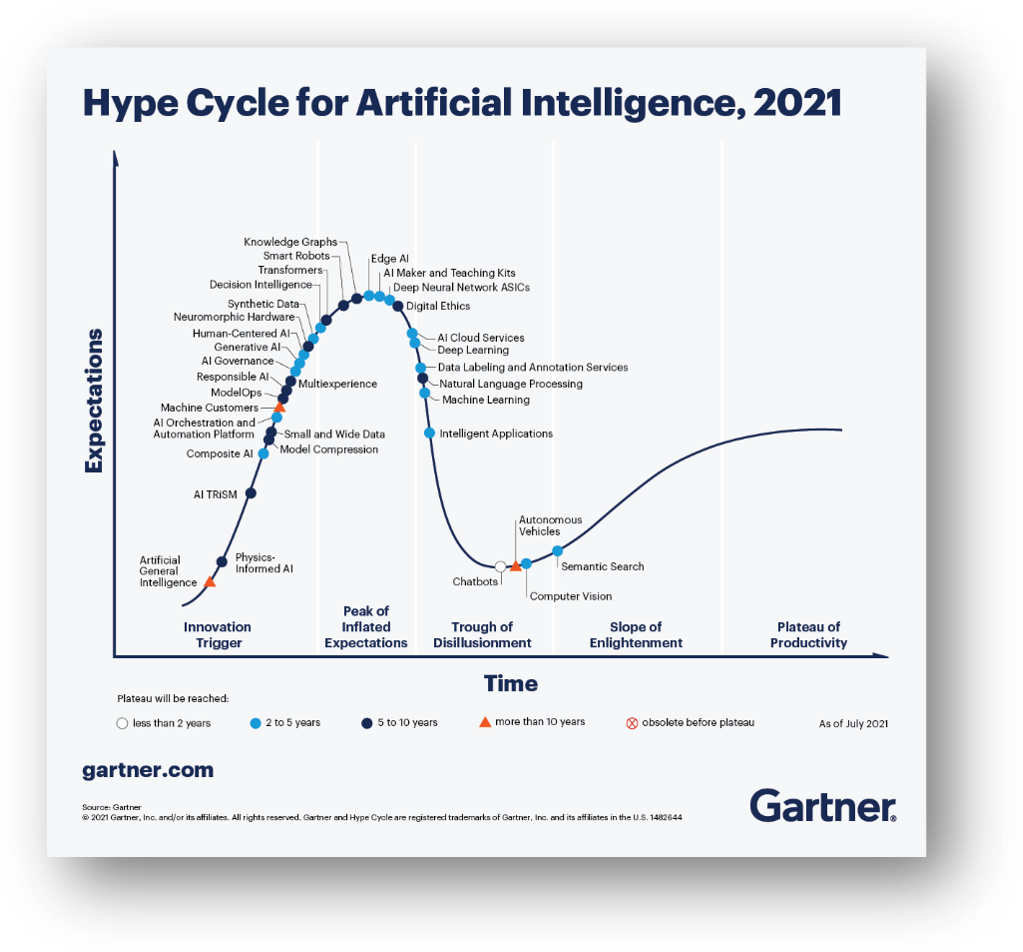
There is a disconnect between the obvious potential for A.I., the marketing claims for A.I. systems, and what is actually delivered. That brings us to the US FDA and post-market surveillance.
In January 2021, FDA published a draft action plan[ii] on AI/ML-based software as a medical device. The plan discusses monitoring of devices from market development through post-market performance. FDA’s stated goal was to allow it reasonable assurance of the safety and effectiveness of such devices.
In fact, the basic idea is sound. By monitoring each device’s performance after clinical installation, one could examine whether the device is in fact performing 1) as expected, 2) across the indicated patient populations, and 3) in conformance with device claims made by the manufacturer. But the devil is in the details.
To monitor performance, one must be able to both transfer evidence of individual device performance to the manufacturer (or to a central repository) and determine whether the device performance is good, bad, or neutral. The former is possible; the latter can be a real challenge.
Telemetry of performance measures only becomes possible if manufacturers design medical devices that are network-connected. Further, by incorporating modern secure communications tools, manufacturers can ensure that appropriate data, measurements and metrics are gathered for post-market analysis – without compromising protected health information (PHI). That is the simpler piece of the puzzle.
Let’s use a particular example to illustrate how difficult it can be to determine whether a device is performing well. Consider detection of a low-incidence disease such as breast cancer, where 1,000 women screened results in 5 cancers being detected, on average. Devices performing this task have been commercially available in the US for nearly 25 years. Is it possible, on a case-by-case basis, to determine if one of these algorithms performed as expected? The answer is no. Suppose that 10% of women are recalled for additional imaging, and 20% of those go for a biopsy. The only definitive way to know if a woman has breast cancer is to sample the suspicious tissue and send the sample to pathology.
In our example, that means that 2% of studies will have a definitive answer. What about the other 98% of cases? Did the algorithm do the right thing on those? Did the practitioner interpret the results correctly (which may not be a reflection on the algorithm)? Was the cancer missed by both algorithm and practitioner?
For a manufacturer to learn the truth about an individual study, that manufacturer must have access to the downstream medical outcome of all the procedures that followed the initial screening mammogram, including diagnostic mammography, possibly ultrasound and/or breast MRI, biopsy, and pathology. The results of all those tests may be stored in different electronic health record (EHR) systems, and they may in fact have been performed by unaffiliated providers with no automated health record exchange. In other words, the records required for complete post-market assessment may simply not be accessible.
Manufacturers sometimes monitor a surrogate endpoint to determine the algorithm performance, such as the number of lesions marked over the last 100 cases, called the “marker rate”. If the marker rate deviates from a factory-established metric such as 100 marks per 100 cases, the manufacturer could determine that something is awry. But as to the question whether the algorithm is “doing the right thing”, the answer may remain elusive.
Thus, manufacturers utilizing AI often rely on anecdotal feedback from clinicians to determine whether their AI is working or not. That method has proven to work in the past, although the feedback loop can be long and slow. Is it good enough? Probably not, so manufacturers must strive to do better with each new algorithm and each new algorithm release.
Will any of that satisfy the FDA’s zeal for post-market surveillance? Probably not. But I would caution FDA to limit their expectations for post-market surveillance to that which is reasonably achievable in a clinical production environment, while encouraging providers of EHR systems and AI manufacturers to work together to solve this problem.
[i] https://www.gartner.com/en/articles/the-4-trends-that-prevail-on-the-gartner-hype-cycle-for-ai-2021